
分子动力学
- 刘十三6136月前发布了课程
 GROMACS分子模拟专题六:TOP-ITP文件介绍
GROMACS分子模拟专题六:TOP-ITP文件介绍由于许多初学者遇到的错误多是由于对ITP、TOP等文件不了解所致,所以本课程详细讲解了GROMACS模拟所需文件ITP、TOP的格式内容。学习之后,可对...
¥120 播放量28 - 刘十三6136月前发布了课程
 GROMACS分子模拟专题五:粗粒化力场建立和模拟仿真
GROMACS分子模拟专题五:粗粒化力场建立和模拟仿真粗粒化分子动力学(Coarse Grained Molecular Dynamics, CGMD)方法是近几年来发展起来的一种可以在介观尺度上描述分子体系演变过程的计算机模拟...
¥359 播放量455 - 刘十三6136月前发布了课程
 GROMACS分子模拟专题四:非标准残基力场构建和高分子itp构建
GROMACS分子模拟专题四:非标准残基力场构建和高分子itp构建GROMACS的pdb2gmx程序可以根据输入的坐标文件产生拓扑文件,但是如果体系中有其不能识别的残基名字,那么就会报错,不能生成itp文件。这种情况...
¥299 播放量863 - 刘十三6136月前发布了课程
 GROMACS分子模拟专题三:质心牵引(伞形采样)模拟仿真计算
GROMACS分子模拟专题三:质心牵引(伞形采样)模拟仿真计算1、计算结合自由能结合能(ΔGbind)可由一系列伞形采样模拟得到的平均力势(PMF, potential of mean force)导出. 在这个过程中会创建一系列初始...
¥299 播放量899 - 刘十三6137月前发布了课程
 分子动力学GROMACS模拟仿真-2:快速生成高分子ITP以及模拟
分子动力学GROMACS模拟仿真-2:快速生成高分子ITP以及模拟教你快速建立高分子ITP以及模拟仿真
¥9 播放量25 - 刘十三6137月前发布了课程
 分子动力学GROMACS模拟仿真基础之创建索引(选择任意基团或原子)
分子动力学GROMACS模拟仿真基础之创建索引(选择任意基团或原子)本次课程为大家讲解在GROMACS里面如何创建任意基团或者原子的索引文件,以至于我们可以使用该索引文件进行任意的数据分析。
¥299 播放量12 - 刘十三6137月前发布了课程
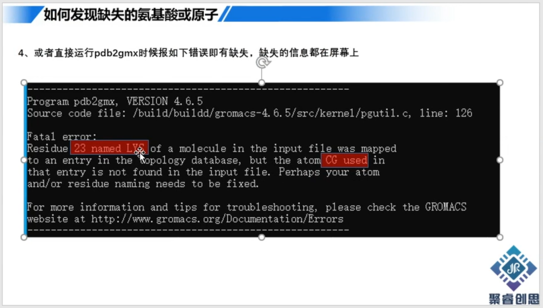 分子动力学GROMACS模拟仿真基础之补全蛋白缺失氨基酸或原子
分子动力学GROMACS模拟仿真基础之补全蛋白缺失氨基酸或原子做蛋白模拟之前,必须对目标蛋白的pdb进行检查。主要是看是否缺失了氨基酸残基或者缺失了氨基酸上面的原子。如果有缺失,那么在模拟之前需要进...
¥199 播放量55 - 刘十三6137月前发布了课程
 分子动力学GROMACS模拟仿真-8:分析命令之盐桥的分析
分子动力学GROMACS模拟仿真-8:分析命令之盐桥的分析通过计算蛋白质之间的盐桥,可以了解蛋白质的稳定性。如何使用gromacs分析盐桥以及注意事项
¥19 播放量28 - 刘十三6137月前发布了课程
 分子动力学GROMACS模拟仿真-3:分析命令之氢键相关性质
分子动力学GROMACS模拟仿真-3:分析命令之氢键相关性质GROMACS分析命令之氢键相关性质讲解
¥19 播放量126 - 刘十三6137月前发布了课程
 分子动力学GROMACS模拟仿真-6:分析命令之径向分布函数函数和配位数
分子动力学GROMACS模拟仿真-6:分析命令之径向分布函数函数和配位数GROMACS分析命令之径向分布函数函数和配位数讲解
¥19 播放量8

