分子动力学模拟:从基础理论到机器学习加速前沿

模拟微观世界,揭示物质奥秘
分子动力学模拟是一种在原子和分子水平上研究体系随时间演化的计算技术,它通过数值求解牛顿运动方程,追踪每个粒子的轨迹,从而揭示物质的微观结构和宏观性质之间的联系。随着计算能力的提升和算法的进步,分子动力学模拟已成为材料科学、生物物理、药物设计等领域不可或缺的研究工具。
分子动力学模拟基础
分子动力学模拟的核心思想相当直观:通过计算每个原子上的作用力,然后根据牛顿第二定律更新原子的位置和速度,如此迭代从而得到体系的演化轨迹。
基本步骤
一个完整的分子动力学模拟通常包含以下步骤:
1.确定起始构型:需要一个能量较低、结构合理的起始三维构型。生物大分子的起始构型主要来自实验数据(如X射线晶体衍射或核磁共振波谱法测定的结构),或根据已知分子结构通过同源建模得到。
2.选用适当的力场和模拟软件:力场决定了原子之间相互作用的数学描述,其选择与模拟结果的准确性 息息相关。软件的选择则往往与所用的力场有关,需考虑所需的算法、运行速度和并行计算能力。
3.构建体系和能量最小化:根据研究对象所处的环境(如气相、水溶液或跨膜环境等)构建模拟体系。初步建立的体系中可能存在局部不合理结构(如原子间隔过近),因此需要进行能量最小化来优化结构。常用的能量最小化方法包括最速下降法和共轭梯度法。
4.平衡过程:体系构建好后,需赋予原子根据特定温度下玻尔兹曼分布的初始速度,并调整速度使体系总体在各方向上的动能之和为零(保证体系无平动位移)。随后在特定系综(如NVT、NPT)下进行模拟,使体系的温度、压强、密度等达到平衡。
5.数据采集:体系平衡后,进行长时间的模拟以采集样本用于分析。记录轨迹(坐标、速度、能量等)的频率需权衡信息完整性和存储开销。
6.结果分析:通过系综平均获得可与实验比较的宏观物理量。分析方法包括计算平均能量及其涨落、分析氢键结构、蛋白质二级结构、结合自由能以及各种动力学性质等。
关键技术要素
在分子动力学模拟中,力场描述了原子间相互作用的势能函数与相关参数,决定了模拟的可靠性。积分算法(如Verlet算法)则用于数值求解运动方程,其稳定性和精度直接影响模拟效果。此外,为了在有限计算资源下模拟宏观体系,通常采用周期性边界条件;在处理长程相互作用时,则需使用快速算法(如Ewald求和)。
传统模拟的挑战与局限
尽管分子动力学模拟功能强大,但也面临诸多挑战:
-时间与空间尺度的限制:传统模拟所能达到的时间尺度和系统尺寸与许多实际物理化学过程(如蛋白质折叠、材料蠕变)相比仍有巨大差距。
-力场精度与适用性:传统经验力场的准确性有限,而高精度的量子力学方法又计算昂贵。
-计算成本高昂:随着原子数目的增加,计算量急剧增长,限制了模拟的规模和时长。
机器学习带来的变革
近年来,机器学习技术的引入正在深刻改变分子动力学模拟的面貌,为解决上述挑战提供了新的途径。
机器学习势函数
机器学习势函数通过机器学习算法从量子力学计算产生的高精度数据中学习势能面,能在保持接近量子力学精度的同时,将计算速度提高数个量级。
国内的渤海大学樊哲勇教授团队开发的GPUMD软件就是一款有竞争力的开源分子动力学模拟软件[1]。GPUMD支持多种经验势以及独具特色的神经演化势(NEP)机器学习势。截至2025年6月,使用GPUMD发表的论文数量连续两年获得接近100%的增长速率,引领同类原子尺度模拟软件。
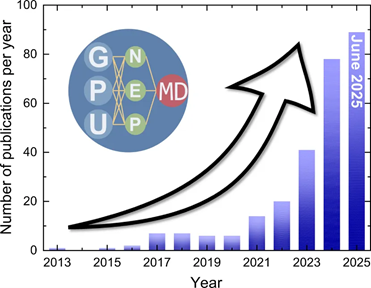
图1每年使用GPUMD发表的论文数(截至2025年6月14日)
GPUMD与NEP的组合兼具速度与精度,已助力众多材料模拟的应用。
新型模拟框架
除了改进势函数,研究者们还在探索全新的模拟范式。
TrajCast是一个基于自回归等变消息传递网络的可迁移且数据高效的框架,它直接更新原子位置和速度,摆脱了传统数值积分对时间步长的约束。
传统分子动力学模拟中,为确保能量守恒和数值稳定性,积分时间步长通常需要很小(如1-2飞秒)。而TrajCast允许的预测时间间隔比传统分子动力学时间步长高出多达30倍,每天可以为包含超过4,000个原子的固体生成超过15纳秒的轨迹数据,显著提升了模拟效率。
DIMOS则是一个端到端可微分的分子模拟框架,用于分子动力学和蒙特卡洛模拟。
这种可微性使得研究者能够通过梯度下降等优化方法,更高效地调整力场参数或模拟参数,例如,该框架已实现了在基于哈密顿蒙特卡洛的马尔可夫链蒙特卡洛模拟中,对提议分布进行端到端优化,使用优化后的模拟参数,比临时选择的参数观察到了3倍的加速。
DIMOS的模块化设计使得经典方法和机器学习方法可以轻松结合成系统的混合描述(ML/MM)。
不确定性量化
机器学习模型在分子动力学中的应用也带来了新的挑战,特别是如何评估和量化这些模型预测的不确定性。
杜克大学的研究者于2025年3月发表了一项工作,提出一个系统无关的概率框架,用于量化基于机器学习势函数的分子动力学模拟中的模型形式不确定性。
这种框架通过随机降阶模型,利用多种机器学习势函数(如NEP和MTP)来编码模型不确定性,并能将这些不确定性传递到宏观量(如离子扩散率),从而增强模拟预测的鲁棒性。
典型应用场景
分子动力学模拟的应用已渗透到多个前沿研究领域:
1.材料热传输行为研究:天津大学封伟教授、香港中文大学(深圳)郑庆彬教授和香港理工大学沈曦教授合作,结合分子动力学模拟与实验,探究了分子界面的热传输行为。
2.药物分子跨膜运输:使用伞形采样算法模拟药物分子穿越细胞膜,为药物设计提供机理insights。
3.能源材料开发:如钠离子电池中的钠硫代磷酸盐系统,机器学习势函数可用于研究其离子扩散行为,不确定性量化则能提高预测的可靠性。
未来展望
分子动力学模拟的未来发展可能集中在以下几个方向:
-电荷自由度的引入:更精确地处理带电体系和化学反应。
-粗粒化模型的开发:在保持准确性的同时,进一步拓展模拟的时间和空间尺度。
-增强采样与时间加速方法:更有效地捕获稀有事件。
-电子哈密顿模型的构造与应用:实现更精确的电子结构计算与分子动力学的结合。
结语
从最初的基础理论探索,到如今与机器学习等前沿技术的深度融合,分子动力学模拟正处于一个激动人心的变革时代。机器学习等新思想的注入,正不断突破传统模拟的界限,让我们能够探索更为复杂、更接近真实的体系,在揭示自然奥秘的道路上不断前行。
参考出处
[1] 渤海大学樊哲勇教授团队|GPUMD 4.0: 基于机器学习势的高性能、多功能材料模拟分子动力学程序包- X-MOL资讯【https://www.x-mol.com/news/935486】
[2] Thiemann F L ,Reschützegger, Thiago, Esposito M ,et al.Force-Free Molecular Dynamics Through Autoregressive Equivariant Networks[J]. 2025.
[3] Christiansen H , Maruyama T , Errica F ,et al.Fast, Modular, and Differentiable Framework for Machine Learning-Enhanced Molecular Simulations[J]. 2025.
[4] Quek A , Ouyang N , Lin H M ,et al.Enhancing robustness in machine-learning-accelerated molecular dynamics: A multi-model nonparametric probabilistic approach[J].Mechanics of Materials, 2025, 202(000).DOI:10.1016/j.mechmat.2024.105237.
免责声明:本文内容基于公开科研资料整理,仅供科普交流使用,不代表任何机构立场。文中提及的研究成果和软件工具均来学者发表的学术文献,读者在科研应用中请以原始文献为准。本文作者不对任何基于本文内容做出的科研决策或投资行为承担责任,如有侵权请联系删除。
我们提供以下仿真方向的技术支持服务:
1、有限元:强度、刚度、变形等结构静力学,振动、冲击、模态、谐响应(如电机振动)、响应谱(如地震)、随机振动、瞬态动力学、显示动力学(如碰撞、跌落、冲击损伤、爆炸冲击波)等结构动力学,屈曲、失稳、冲压、钣金成型,结构疲劳;流体仿真、传热仿真、电子产品热仿真、电磁仿真、凝固与融化、仿真、代做
2、多物理场耦合:热应力、热变形约束、涡激振动、液晃、血流冲击、感应加热、微波消融、金属凝固、焊接熔池、激光加工
3、物理光学/几何光学:光波导、硅基芯片、微纳光学、光纤传感、超材料和超表面设计、光子晶体、光力/扭矩计算;光学成像、照明设计、激光系统
4、分子动力学:同源建模、分子对接、蛋白对接、结合能、氢键作用、范德华力、相互作用、结构预测、蛋白筛选、药物筛选、构象、RMSF、高分子、大分子、团簇、溶液体系、分子轨道能量、运动轨迹、材料拉伸
5、程序开发:图像处理、机器学习、数值计算、信号处理、模糊PID控制、电子电力、控制器设计、simulink仿真、GUI界面、算法优化、曲线拟合、金融建模、统计分析、数字孪生
6、.....



